


Sterile Filter Validation
1 Background
Although it is widely recognized that filter validation studies must be performed for aseptic processing, there is no specific guidance that categorically states when filter validation studies should be performed. Consequently, the decision when to perform filter validation is determined largely by the end-user. Risk mitigation is central to quality by design (QbD) and quality risk management (QRM), and increasingly, ways are sought to build quality into production steps, to reduce risk to the patient [1-3]. With respect to aseptic processing, this includes a thorough understanding of the design space for sterile filtration.
Filter validation studies are typically performed during Phase 3 clinical trials, with the final filter validation documentation submitted when the new drug application (NDA) is filed [4-8]. Part of the filter validation testing requires process-specific bacterial challenge tests to be conducted. This assesses the ability of the sterilizing grade filter to product a sterile fluid, even under the worst-case conditions of the process.
As biopharmaceutical processes have become increasingly sophisticated, so too have the requirements for performing process-specific bacterial retention studies. This can often mean that innovative test designs are required to meet the regulatory requirements, while offering the flexibility to simulate the end users process. Further, with the introduction of novel nanoparticle drug delivery systems, such as liposomes and emulsions, accurately simulating processing conditions has significantly increased in complexity.
Pall has communicated extensively in recent years of new challenges surrounding the sterile filtration of so called, ‘high risk fluids’. Occurrences of failed bacterial retention studies (i.e. penetration of bacteria through the sterilizing grade filter when tested under the process conditions) are very rare. However, when penetration has been observed, Pall has reported that low surface tension fluids (typically < 68 dynes/cm2), specifically surfactant solutions, liposomes, lipids, emulsions and lipid-like solutions pose the greatest risk of allowing bacterial penetration through 0.2 µm rated, sterilizing grade filters [9-11].
Therefore, Pall recommends that a risk assessment is performed as early as possible, but no later than during Phase 2 clinical trials, to review any potential risks for sterility assurance. If the risk is deemed sufficiently low, then the end-users may elect to perform filter validation studies in Phase 3. For end-users who proactively want to apply QbD into sterile filtration at Phase 2, then Pall can assist with screening studies as part of the sterility optimization by assessment of risk (SOAR) Program.
The purpose of this document is to outline Pall’s recommendations for applying the principles of risk management to sterility assurance by aseptic processing.
2 What is the Purpose of the SOAR Program?
The purpose of Pall’s SOAR program is to evaluate the risk to sterility of any given set of processing conditions prior to performing formal filter validation studies. This is important, as it allows the end user to fully explore the proposed filter design space at the time of filter selection. While not every process fluid or sterile application may need this detailed level of understanding, there are significant benefits to considering this during Phase 2 clinical trials. The program is split into the three main areas, as shown in Figure 1. Figure 1 Elements of Pall’s SOAR Program for Evaluation of Sterility Assurance.
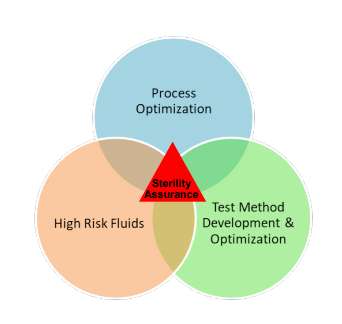
3 Process Optimization
The purpose of this program is to allow the end user to assess any potential risk to sterility assurance caused by the intended manufacturing process. For example, for small batch volumes, the process may involve implementing a filter blow-down step with compressed air post-filtration to maximize product recovery. Therefore, it may be appropriate to use this program to determine appropriate pressures and durations to optimize product recovery, as well as assess any potential risk to sterility of the final product. By conducting such trials earlier in product development, the end user can communicate their risk mitigation measures to the regulatory authorities in their annual reports, decreasing the amount of work required for NDA submission.
3.1 Test Method Development and Optimization
This program allows the validation laboratory to design and qualify new test set-ups which may be required for complex processing operations. This may be required for customers who request high operating parameters that may be difficult to obtain in a scaled down laboratory test. Recognition of these types of projects earlier on in product development ultimately increases the speed at which the final filter validation work can be completed. During the test method development, different configurations may be used until a suitable scaled down model is developed and qualified that achieves the requested parameters. Sterility Assurance Filtration. Separation. Solution.
3.2 High Risk Fluids
Use of nanoparticles as a drug delivery system is a rapidly growing market. Many drugs that would otherwise be unavailable for patients (for example, due to stability, solubility or toxicity issues) can safely be administered by incorporating the active product ingredient (API) into liposomes or other lipid-derived delivery systems. Liposomes and similar drug delivery systems cannot be terminally sterilized, and therefore, sterile filtration is a critical component in the manufacturing process. However, it is now recognized that these fluids present some unique challenges for sterile filtration. One of the biggest concerns with sterile filtration of nanoparticles is the size distribution of the particles. Ideally, there is a narrow size distribution which is reproducible between different batches. This then allows a suitable sterilizing grade filter configuration to be selected based on maximum product recovery and volume throughput. Once a suitable sterilizing grade filter has been selected, Pall advises performing screening studies for bacterial retention as early as possible. Because of potential differences between batches while product development is ongoing, for the purposes of filter validation, this allows the filter design space to be evaluated early on, and any limiting factors to be understood well in advance of NDA submission. Ultimately, this can help determine the upper limits of the manufacturing process and build in safety factors that are appropriate based on actual data.
4 Benefits of the SOAR Program
Early implementation of QbD and QRM into a process is a regulatory expectation, and risk mitigation is being reviewed with increased scrutiny. Further, with respect to the cost of developing a new drug product, it is desirable to reduce the time to market as much as possible. With increasingly complex biopharmaceutical manufacturing operations and drug delivery systems, the traditional approach to sterile filtration and subsequent filter validation performed as a one-time event may not be sufficiently robust to accommodate the current regulatory expectations.
5 References
1. ICH Quality Risk Management Q9, November 2005.
2. Guidance for Industry Q10 Pharmaceutical Quality System U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) April 2009.
3. Guidance for Industry Quality Systems Approach to Pharmaceutical cGMP Regulations. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Center for Veterinary Medicine (CVM) Office of Regulatory Affairs (ORA) September 2006.
4. Guidance for Industry CGMP for Phase 1 Investigational Drugs, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory Affairs (ORA), July 2008.
5. EU Guidelines to Good Manufacturing Practice. Medicinal Products for Human and Veterinary Use, Annex 13. Investigational Medicinal Products, February 2010.
6. Guidance for Industry INDs for Phase 2 and Phase 3 Studies. Chemistry, Manufacturing and Controls Information. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), May 2003.
7. Guidance for Industry Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products. Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM), May 2003.
8. Guidance for Industry Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products. Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM), May 2003.
9. Onraedt, A.; Folmsbee, M.; Kumar, A. & Martin, J. (2010), Sterilizing filtration of adjuvanted vaccines: Ensuring successful filter qualification. BioPharm International Supplement. October.
10. Folmsbee M. & Moussourakis M. (2012). Sterilizing filtration of liposome and related lipid-containing solutions: enhancing successful filter qualification. PDA J Pharm Sci Technol., 66(2): 161-167. 11. Folmsbee, M. (2015). Evaluation of the effect of the volume throughput and maximum flux of low surfacetension fluids on bacterial penetration of 0.2 micron rated filters during process-specific filter validation testing, PDA J Pharm Sci Technol., 69: 307-316.
Doc ref: RPSCSFV_02